Файл: Динамическая вязкость. Закон Ньютона вязкостного трения. Кинетическая теория вязкости по дисциплине.docx
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 09.02.2024
Просмотров: 13
Скачиваний: 0
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.

А.
Наиболее просто этот результат может быть получен следующим образом. Будем рассматривать одну из частиц жидкости как макроскопический шарик с радиусом α и определим сопротивление F, которое он испытывает со стороны окружающей жидкости при движении относительно нее со средней скоростью υ по известной формуле Стокса
F=6παηυ (7)
Переписывая эту формулу в виде υ=uF, мы видим, что подвижность рассматриваемой частицы u может быть выражена через коэффициент вязкости η формулой
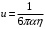 (8)
(8)
С другой стороны, согласно соотношению Эйнштейна (справедливому при не слишком больших значениях силы F)
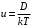 (9)
(9)
Комбинируя эти формулы получаем
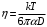 (10)
(10)
Возвращаясь к "микроскопической" точке зрения и подразумевая под D коэффициент самодиффузии рассматриваемой жидкости, имеем
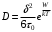 (11)
(11)
При подстановке этого выражения в предыдущую формулу она принимает вид
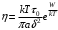 (12)
(12)
то есть сводится к формуле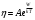 , если положить в ней
, если положить в ней
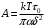 (13)
(13)
Формула (12) очень точно описывает зависимость вязкости не только простых, но и более сложных жидкостей от температуры при постоянном внешнем давлении. Экспериментальные значения коэффициента А оказываются, однако, примерно в 100 – 1000 раз меньше теоретического значения. Последнее обстоятельство объясняется той же причиной, что и кажущееся несовпадение предэкспоненциального множителя в выражении (11) для коэффициента диффузии с теоретическим значением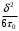 , а именно уменьшением энергии W с повышением температуры вследствие теплового расширения жидкости при постоянном давлении. Полагая, согласно формуле
, а именно уменьшением энергии W с повышением температуры вследствие теплового расширения жидкости при постоянном давлении. Полагая, согласно формуле
W=W0-γkT(14),
для коэффициента диффузии получается выражение
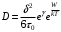 (15)
(15)
и соответственно этому прежнее выражение (6) для коэффициента вязкости со значением А, уменьшенным в eγ раз, то есть
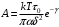
(16)
Для приведения теоретических значений в согласие с экспериментальными следует положить eγ =100, то есть γ=5, что соответствует уменьшению энергии разрыхления W примерно на 10кал/моль при повышении Т на 10. Соответственно этому, полученные выше значения τ нужно увеличить примерно в 100 раз
Если учитывать зависимость W не только от температуры, но и от давления, согласно формулам W = W0–β(V-V0) и V-V0=-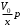 , то для коэффициента А получается экспоненциальная зависимость от давления
, то для коэффициента А получается экспоненциальная зависимость от давления
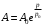 (17), где
(17), где 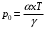
Последняя величина представляет собой давление, при котором, согласно нашей теории, вязкость должна увеличиваться в е=2,7 раз. Полагая здесь α=10-5 и х=1012 дин/см2, получаем при γ=5 и Т=3000:
р0=109 дин/см2=1000кг/см2, т.е. 1000 атм
Отсюда следует, что при давлении р=10000 атм. вязкость жидкости должна увеличиваться по сравнению со своим нормальным значением при той же (комнатной) температуре примерно в е10=104 раз. Этот результат дает несколько преувеличенное представление о порядке величины влияния, оказываемого давлением на вязкость жидкостей. Для разных жидкостей это влияние может быть, одинаково, весьма различным в зависимости от соответствующего значения р0. Следует также иметь в виду, что распространение формулы (17) на какие угодно высокие давления недопустимо потому, что эта формула основана на предположении о линейной зависимости энергии разрыхления W от объема и линейной зависимости последнего от давления; при сверхвысоких давлениях эти зависимости могут иметь более сложный характер.
Сам Я.И. Френкель признает, что вывод формул (12) и (13) страдает некоторым формализмом, не раскрывая конкретным образом того механизма, который лежит в основе процесса вязкого течения жидкости и определяет величину коэффициента вязкости. Этим объясняется тот факт, что разные авторы, занимавшиеся вопросом о вязкости жидкости, в частности, Андраде, давший в 1928 году теорию скорее качественного, чем количественного характера, не заметил этого сходства. Не уяснил себе сущности теории Френкеля и Эйринг, который развил ее в 1938 году с помощью своего метода "промежуточного комплекса", используя ту же самую идею перехода частиц из одного положения равновесия в другое.
Проведя анализ молекулярно-кинетического механизма, лежащего в основе течения жидкостей, Френкель подошел к вопросу об определении коэффициента вязкости другим путем, намеченным еще Максвеллом. Прежние выражения для коэффициента вязкости оказались эквивалентны новым, полученным на основе Максвелловской релаксационной теории упругости.
Если теперь вместо частицы самой жидкости рассматривать частицы какого-нибудь растворенного в ней постороннего вещества, то, применяя формулу Стокса, получим для подвижности этой «чужой» частицы значение того же порядка величины, что и для «своей» (поскольку различие между радиусами частиц невелико). Этот результат непосредственно подтверждается в случае ионов в водных (или иных) растворах электролитов, по электропроводности которых может быть вычислена подвижность этих ионов. При этом оказывается, что подвижность различных ионов имеет одинаковый порядок величины и одинаковый температурный ход, соответствующий температурной зависимости вязкости растворителя.
И коэффициенты диффузии их D=ukTдолжны быть также приблизительно одинаковыми, то есть иметь такой же порядок величины и такой же температурный ход, что и коэффициент самодиффузии этой жидкости. Этот вывод находится в полном согласии с экспериментальными данными. В случае жидкостей коэффициенты диффузии различных растворенных в ней веществ – твердых, жидких и газообразных – имеют сходные значения, лежащие при комнатной температуре в пределах от нескольких десятых до нескольких единиц квадратных сантиметров в сутки.
Коэффициенты диффузии в жидкостях, как правило, мало зависят от природы растворителя. Так, например, коэффициенты диффузии свинца, кадмия и золота в ртути при различных температурах, лежащих в
интервале от комнатной до 500°С, изменяются в пределах всего лишь
0,72 – 3,18 см2/сутки; заметим для сравнения, что коэффициенты диффузии золота в расплавленном свинце и олове при 500°С равны
3,19 и 4,65 см2/сутки, а коэффициенты диффузии различных органических веществ в органических жидкостях при 20°С колеблются в пределах
0,4 – 2,6 см2/сутки.
Только в случае очень вязких жидкостей коэффициенты диффузии различных растворенных веществ могут принимать существенно отличные друг от друга значения, изменяясь обратно пропорционально вязкости при изменении растворителя, а также его температуры.
В формуле (15), определяющей коэффициент диффузии
, множитель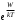 должен быть гораздо ближе к 1 и изменяться гораздо меньше с температурой.
должен быть гораздо ближе к 1 и изменяться гораздо меньше с температурой.
Этим объясняется тот факт, что коэффициенты диффузии разных веществ в жидкостях во много раз (на несколько порядков величины) больше, чем в твердых телах и гораздо менее чувствительны к изменению температуры, а также природы растворителя или растворенного вещества.
Приблизительная одинаковость энергии разрыхления W для различных веществ, растворенных в одной и той же жидкости, и (приблизительное) совпадение ее с энергией активации, определяющей температурную зависимость коэффициента вязкости растворителя, могут быть объяснены следующим образом.
1. Если растворенное вещество состоит из молекул, размеры которых очень велики по сравнению с размерами молекул растворителя (например, растворы сахара или различных органических красителей в воде), то представление о тепловом движении частиц жидкости может применяться лишь к молекулам растворителя, а не к молекулам растворенных веществ. Так же как и частицы коллоидных растворов, эти «макромолекулы» совершают броуновское движение, которое имеет мало общего с колебаниями около временных положений равновесия и перескоками в соседние положения равновесия. Представление о подобных положениях не имеет в этом случае никакого смысла. Поэтому введение энергии активации W, характеризующей длительность пребывания подобной макромолекулы в одном и том же положении равновесия (по формуле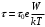 ), также не имеет смысла. Соответственно этому утрачивает смысл и формула (11) для коэффициента диффузии, если подразумевать в ней под δ элементарное перемещение макромолекулы, а под τ0 – период ее колебаний около равновесного положения. Формула же
), также не имеет смысла. Соответственно этому утрачивает смысл и формула (11) для коэффициента диффузии, если подразумевать в ней под δ элементарное перемещение макромолекулы, а под τ0 – период ее колебаний около равновесного положения. Формула же
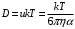 становится, наоборот, тем более точной, чем больше линейные размеры макромолекулы α. Полагая в этой формуле
становится, наоборот, тем более точной, чем больше линейные размеры макромолекулы α. Полагая в этой формуле 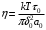 , согласно , где величины τ0,a0, δ0, и W0 относятся к молекулам растворителя, получаем для коэффициента диффузии выражение
, согласно , где величины τ0,a0, δ0, и W0 относятся к молекулам растворителя, получаем для коэффициента диффузии выражение
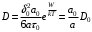 (18)
(18)
которое как в отношении характера температурной зависимости, так и в отношении порядка величины предэкспоненциального множителя находится в хорошем согласии с данными опыта.
2. Если молекулы растворенного вещества по своим размерам сравнимы с молекулами растворителя, то для них должно оставаться в силе обычное представление о тепловом движении в жидкостях и вытекающая из этих представлений формула (11). При этом энергия активации W должна, казалось бы, определяться взаимодействием молекулы растворенного вещества с окружающими ее молекулами растворителя и потому должна была бы, вообще говоря, существенно отличаться от энергии активации W0, характеризующей растворитель. Энергия W во всех случаях оказывается близкой к W0.
Это обстоятельство можно объяснить двумя способами:
а) Можно представить, что при перемещении маленьких молекул растворенного вещества окружающие молекулы растворителя увлекаются примерно таким же образом, как и при перемещении макромолекул. «Увлечение» молекул растворителя молекулой растворенного вещества имеет совершенно такой же характер, как и взаимное увлечение молекул растворителя в случае самодиффузии. При таких условиях энергия активации W, относясь не к одной лишь центральной частице (своей или чужой), а ко всему комплексу частиц, так или иначе участвующих в элементарном перемещении, не может полностью определяться взаимодействием растворенной частицы с частицами растворителя, но должна существенным образом зависеть от взаимодействия последних друг с другом. Ясно, что при таких условиях W не может существенно отличаться от W0.
Рассмотрим перемещения группы частиц в другом направлении так, чтобы рассматриваемая (центральная» частица (своя или чужая) была не «ведущей», а «ведомой», подобно тому как это имеет место в дырочном механизме диффузии в кристаллах. Представим себе, что перемещение рассматриваемой частицы из исходного положения равновесия в соседнее имеет не активный, а пассивный характер, то есть обусловливается не случайным увеличением кинетической энергии рассматриваемой частицы при неизменном расположении окружающих, а образованием в непосредственной близости к данной частице микрополости (дырки), в которую она может перейти практически без всякой энергии активации, после чего дырка, оставленная ею на прежнем месте, захлопывается. С этой точки зрения энергию активации
Наиболее просто этот результат может быть получен следующим образом. Будем рассматривать одну из частиц жидкости как макроскопический шарик с радиусом α и определим сопротивление F, которое он испытывает со стороны окружающей жидкости при движении относительно нее со средней скоростью υ по известной формуле Стокса
F=6παηυ (7)
Переписывая эту формулу в виде υ=uF, мы видим, что подвижность рассматриваемой частицы u может быть выражена через коэффициент вязкости η формулой
(8)С другой стороны, согласно соотношению Эйнштейна (справедливому при не слишком больших значениях силы F)
(9)Комбинируя эти формулы получаем
(10)Возвращаясь к "микроскопической" точке зрения и подразумевая под D коэффициент самодиффузии рассматриваемой жидкости, имеем
(11)При подстановке этого выражения в предыдущую формулу она принимает вид
(12)то есть сводится к формуле
, если положить в ней (13)Формула (12) очень точно описывает зависимость вязкости не только простых, но и более сложных жидкостей от температуры при постоянном внешнем давлении. Экспериментальные значения коэффициента А оказываются, однако, примерно в 100 – 1000 раз меньше теоретического значения. Последнее обстоятельство объясняется той же причиной, что и кажущееся несовпадение предэкспоненциального множителя в выражении (11) для коэффициента диффузии с теоретическим значением
, а именно уменьшением энергии W с повышением температуры вследствие теплового расширения жидкости при постоянном давлении. Полагая, согласно формулеW=W0-γkT(14),
для коэффициента диффузии получается выражение
(15)и соответственно этому прежнее выражение (6) для коэффициента вязкости со значением А, уменьшенным в eγ раз, то есть
(16)
Для приведения теоретических значений в согласие с экспериментальными следует положить eγ =100, то есть γ=5, что соответствует уменьшению энергии разрыхления W примерно на 10кал/моль при повышении Т на 10. Соответственно этому, полученные выше значения τ нужно увеличить примерно в 100 раз
Если учитывать зависимость W не только от температуры, но и от давления, согласно формулам W = W0–β(V-V0) и V-V0=-
, то для коэффициента А получается экспоненциальная зависимость от давления (17), где Последняя величина представляет собой давление, при котором, согласно нашей теории, вязкость должна увеличиваться в е=2,7 раз. Полагая здесь α=10-5 и х=1012 дин/см2, получаем при γ=5 и Т=3000:
р0=109 дин/см2=1000кг/см2, т.е. 1000 атм
Отсюда следует, что при давлении р=10000 атм. вязкость жидкости должна увеличиваться по сравнению со своим нормальным значением при той же (комнатной) температуре примерно в е10=104 раз. Этот результат дает несколько преувеличенное представление о порядке величины влияния, оказываемого давлением на вязкость жидкостей. Для разных жидкостей это влияние может быть, одинаково, весьма различным в зависимости от соответствующего значения р0. Следует также иметь в виду, что распространение формулы (17) на какие угодно высокие давления недопустимо потому, что эта формула основана на предположении о линейной зависимости энергии разрыхления W от объема и линейной зависимости последнего от давления; при сверхвысоких давлениях эти зависимости могут иметь более сложный характер.
Сам Я.И. Френкель признает, что вывод формул (12) и (13) страдает некоторым формализмом, не раскрывая конкретным образом того механизма, который лежит в основе процесса вязкого течения жидкости и определяет величину коэффициента вязкости. Этим объясняется тот факт, что разные авторы, занимавшиеся вопросом о вязкости жидкости, в частности, Андраде, давший в 1928 году теорию скорее качественного, чем количественного характера, не заметил этого сходства. Не уяснил себе сущности теории Френкеля и Эйринг, который развил ее в 1938 году с помощью своего метода "промежуточного комплекса", используя ту же самую идею перехода частиц из одного положения равновесия в другое.
Проведя анализ молекулярно-кинетического механизма, лежащего в основе течения жидкостей, Френкель подошел к вопросу об определении коэффициента вязкости другим путем, намеченным еще Максвеллом. Прежние выражения для коэффициента вязкости оказались эквивалентны новым, полученным на основе Максвелловской релаксационной теории упругости.
Если теперь вместо частицы самой жидкости рассматривать частицы какого-нибудь растворенного в ней постороннего вещества, то, применяя формулу Стокса, получим для подвижности этой «чужой» частицы значение того же порядка величины, что и для «своей» (поскольку различие между радиусами частиц невелико). Этот результат непосредственно подтверждается в случае ионов в водных (или иных) растворах электролитов, по электропроводности которых может быть вычислена подвижность этих ионов. При этом оказывается, что подвижность различных ионов имеет одинаковый порядок величины и одинаковый температурный ход, соответствующий температурной зависимости вязкости растворителя.
И коэффициенты диффузии их D=ukTдолжны быть также приблизительно одинаковыми, то есть иметь такой же порядок величины и такой же температурный ход, что и коэффициент самодиффузии этой жидкости. Этот вывод находится в полном согласии с экспериментальными данными. В случае жидкостей коэффициенты диффузии различных растворенных в ней веществ – твердых, жидких и газообразных – имеют сходные значения, лежащие при комнатной температуре в пределах от нескольких десятых до нескольких единиц квадратных сантиметров в сутки.
Коэффициенты диффузии в жидкостях, как правило, мало зависят от природы растворителя. Так, например, коэффициенты диффузии свинца, кадмия и золота в ртути при различных температурах, лежащих в
интервале от комнатной до 500°С, изменяются в пределах всего лишь
0,72 – 3,18 см2/сутки; заметим для сравнения, что коэффициенты диффузии золота в расплавленном свинце и олове при 500°С равны
3,19 и 4,65 см2/сутки, а коэффициенты диффузии различных органических веществ в органических жидкостях при 20°С колеблются в пределах
0,4 – 2,6 см2/сутки.
Только в случае очень вязких жидкостей коэффициенты диффузии различных растворенных веществ могут принимать существенно отличные друг от друга значения, изменяясь обратно пропорционально вязкости при изменении растворителя, а также его температуры.
В формуле (15), определяющей коэффициент диффузии
, множитель
должен быть гораздо ближе к 1 и изменяться гораздо меньше с температурой.Этим объясняется тот факт, что коэффициенты диффузии разных веществ в жидкостях во много раз (на несколько порядков величины) больше, чем в твердых телах и гораздо менее чувствительны к изменению температуры, а также природы растворителя или растворенного вещества.
Приблизительная одинаковость энергии разрыхления W для различных веществ, растворенных в одной и той же жидкости, и (приблизительное) совпадение ее с энергией активации, определяющей температурную зависимость коэффициента вязкости растворителя, могут быть объяснены следующим образом.
1. Если растворенное вещество состоит из молекул, размеры которых очень велики по сравнению с размерами молекул растворителя (например, растворы сахара или различных органических красителей в воде), то представление о тепловом движении частиц жидкости может применяться лишь к молекулам растворителя, а не к молекулам растворенных веществ. Так же как и частицы коллоидных растворов, эти «макромолекулы» совершают броуновское движение, которое имеет мало общего с колебаниями около временных положений равновесия и перескоками в соседние положения равновесия. Представление о подобных положениях не имеет в этом случае никакого смысла. Поэтому введение энергии активации W, характеризующей длительность пребывания подобной макромолекулы в одном и том же положении равновесия (по формуле
), также не имеет смысла. Соответственно этому утрачивает смысл и формула (11) для коэффициента диффузии, если подразумевать в ней под δ элементарное перемещение макромолекулы, а под τ0 – период ее колебаний около равновесного положения. Формула же становится, наоборот, тем более точной, чем больше линейные размеры макромолекулы α. Полагая в этой формуле , согласно , где величины τ0,a0, δ0, и W0 относятся к молекулам растворителя, получаем для коэффициента диффузии выражение (18)которое как в отношении характера температурной зависимости, так и в отношении порядка величины предэкспоненциального множителя находится в хорошем согласии с данными опыта.
2. Если молекулы растворенного вещества по своим размерам сравнимы с молекулами растворителя, то для них должно оставаться в силе обычное представление о тепловом движении в жидкостях и вытекающая из этих представлений формула (11). При этом энергия активации W должна, казалось бы, определяться взаимодействием молекулы растворенного вещества с окружающими ее молекулами растворителя и потому должна была бы, вообще говоря, существенно отличаться от энергии активации W0, характеризующей растворитель. Энергия W во всех случаях оказывается близкой к W0.
Это обстоятельство можно объяснить двумя способами:
а) Можно представить, что при перемещении маленьких молекул растворенного вещества окружающие молекулы растворителя увлекаются примерно таким же образом, как и при перемещении макромолекул. «Увлечение» молекул растворителя молекулой растворенного вещества имеет совершенно такой же характер, как и взаимное увлечение молекул растворителя в случае самодиффузии. При таких условиях энергия активации W, относясь не к одной лишь центральной частице (своей или чужой), а ко всему комплексу частиц, так или иначе участвующих в элементарном перемещении, не может полностью определяться взаимодействием растворенной частицы с частицами растворителя, но должна существенным образом зависеть от взаимодействия последних друг с другом. Ясно, что при таких условиях W не может существенно отличаться от W0.
Рассмотрим перемещения группы частиц в другом направлении так, чтобы рассматриваемая (центральная» частица (своя или чужая) была не «ведущей», а «ведомой», подобно тому как это имеет место в дырочном механизме диффузии в кристаллах. Представим себе, что перемещение рассматриваемой частицы из исходного положения равновесия в соседнее имеет не активный, а пассивный характер, то есть обусловливается не случайным увеличением кинетической энергии рассматриваемой частицы при неизменном расположении окружающих, а образованием в непосредственной близости к данной частице микрополости (дырки), в которую она может перейти практически без всякой энергии активации, после чего дырка, оставленная ею на прежнем месте, захлопывается. С этой точки зрения энергию активации

