Файл: Выделить основные аспекты диагностики и лечения миастении и миастенических синдромов.docx
ВУЗ: Не указан
Категория: Не указан
Дисциплина: Не указана
Добавлен: 18.03.2024
Просмотров: 39
Скачиваний: 0
ВНИМАНИЕ! Если данный файл нарушает Ваши авторские права, то обязательно сообщите нам.

3)Назначение цефалоспоринов при первых признаках пневмонии.
4)Адекватное энтеральное или — при бульбарных нарушениях — парентеральное питание.
Алгоритм экстренных мероприятий при миастеническом кризе:
1)Прозерин в/в по 1–2 мл 0,05% раствора; при необходимости 2–3 раза в сутки.
2)Иммуноглобулин в/в в суточной дозе 400 мг.
3)При неэффективности перечисленных мероприятий дополнительно назначается преднизолон в суточной дозе 100 мг.
4)Для профилактики пневмонии — цефалоспорины III и IV поколений (цефотаксим, цефтриаксон, цефтазидим, цефметазол, цефпирон и др.).
5)Для купирования возбуждения — в/в или в/м 1 мл 0,5% раствора галоперидола (до 20 мг/сут).
Алгоритм экстренных мероприятий при холинергическом кризе:
1)Немедленная отмена антихолинэстеразных препаратов.
2)Внутримышечное или подкожное — в зависимости от тяжести состояния больного — введение 0,5–1,0 мл 0,1% раствора атропина. Инъекции могут при необходимости повторяться с интервалом в 1–2 ч до появления сухости во рту.
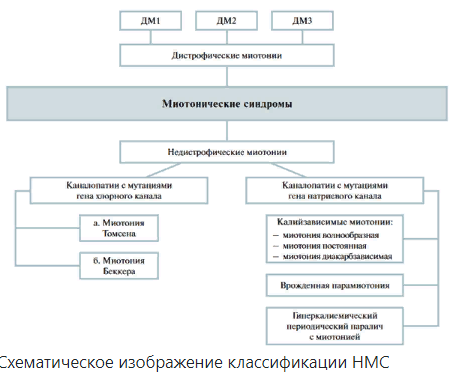
ОПРЕДЕЛЕНИЕ НАСЛЕДСТВЕННЫХ МИОТОНИЧЕСКИХ СИНДРОМОВ. КЛАССИФИКАЦИЯ.
Наследственные миотонические синдромы(НМС)- группа генетически гетерогенных заболеваний ионных каналов хлора и натрия (каналопатии), характеризующиеся повышенной возбудимостью мембраны мышечных волокон, проявляющиеся миотоническими феноменами с постоянной или транзиторной слабостью скелетной мускулатуры. В современной классификации НМС представлены различными генетическими формами дистрофических (ДМ) и недистрофических миотоний (НДМ) (Mankodi A.2008г)
ДИСТРОФИЧЕСКАЯ МИОТОНИЯ. ГЕНЕТИЧЕСКИЕ АСПЕКТЫ, ЭТИОПАТОГЕНЕЗ, КЛАССИФИКАЦИЯ
Дистрофическая миотония (ДМ) (сongenital myotonic dystrophy) является наследственным мультисистемным заболеванием, при котором мутация затрагивает развитие и функционирование различных органов и тканей: гладкой и скелетной мышечной ткани, сердца, органа зрения (глаза), головного мозга. Клиническая картина дистрофической миотонии складывается из 3 синдромов: миотонического, дистрофического и синдрома вегетативно-трофических нарушений. Ключевая особенность заболевания — сочетание миотонии, которая характеризуется отсроченным расслаблением после мышечного сокращения, и прогрессирующей мышечной слабости, дистрофии (атрофии). До 1994 года дистрофическая миотония считалась однородным заболеванием. Однако в последние годы после идентификации различных мутаций при сходной клинической симптоматике, напоминающей дистрофическую миотонию, было показано, что это гетерогенное заболевание, представленное тремя подтипами: дистрофическая миотония 1 типа (мутация 19q13.3), дистрофическая миотония 2 типа (мутация 3q21) и дистрофическая миотония 3 типа (мутация 15q21-q24). Распространенность дистрофической миотонии 1 типа в больших популяциях около 1:8000 , а дистрофической миотонии 2 и 3 типов в настоящее время недостаточно изучена.
В отличие от большинства других наследственных нервно-мышечных заболеваний (и от других форм миотоний, в частности) клинические проявления дистрофической миотонии вариабельны и могут различаться от человека к человеку даже в пределах одной семьи. Больные с дистрофической миотонией могут сталкиваться с большим числом разнообразных (необычных для миотонии) проблем, к которым можно отнести низкий уровень жизненной активности, пассивность, депрессию, облысение, нарушения со стороны желудочно-кишечного тракта, сексуальные проблемы, что в ряде случаев приводит к возникновению недопонимания и даже к конфликтным ситуациям между пациентом и лечащим врачом. Следует помнить, что пассивное поведение пациента является по большей мере реальным проявлением данного заболевания, а не желанием человека.
КЛАССИФИКАЦИЯ
По локализации мутированного гена:
1. дистрофическая миотония 1 типа (мутация 19q13.3) DM1, миотония Штейнерта — Баттена — Куршманна (Steinert — Batten — Curschmann myotonic dystrophy), атрофическая миотония Россолимо;
2. дистрофическая миотония 2 типа (мутация 3q21) DM2;
3. дистрофическая миотония 3 типа (мутация 15q21-q24) DM3;
Классификация дистрофической миотонии 1 по возрасту дебюта заболевания:
1. конгенитальная дистрофическая миотония(врожденная, congenital myotonic dystrophy CmyD) — с характерной клинической симптоматикой сразу при рождении ребенка и серьезным прогнозом (не описана при DM2 и DM3);
2. ювенильная дистрофическая миотония(juvenile DM)— возраст дебюта заболевания от 1 года до подросткового возраста;
3. дистрофическая миотония взрослых(adult DM) — с дебютом у индивидуумов старше 20, но моложе 40 лет;
4. —дистрофическая миотония с поздним дебютом(DM with late onset) — у индивидуумов старше 40 лет и с более легким течением.
ГЕНЕТИКА
При DM1 мутация заключается в экспансии тринуклеотидного повтора ЦТГ (Цитозин-Тимин-Гуадинин) (CTG) в протеинкиназном гене 19-й хромосомы (19q13.3). В норме число CTG-повторов варьирует от 5 до 37, а при DM1 увеличивается от 38 до 2000 и более; наиболее тяжелое течение, ранний дебют и серьезный прогноз заболевания отмечаются у индивидуумов с числом тринуклеотидных повторов, равным 3000 и более.
Продуктом гена является белок миотонинпротеинкиназа ( dystrophy myotonic protein kinase — DMPK) . Этот белок является тканеспецифичным. В настоящее время выделяют 6 подтипов DMPK, которые локализуются в различных тканях: в скелетной и гладкой мышечной ткани, в миокарде (волокна Пуркинье, вставочные диски кардиомиоцитов), в центральной нервной системе (на апикальной мембране эпендимы, plexus choroideus, в синапсах мозжечка, гиппокампе, продолговатом и среднем мозге), фибробластах, лимфоцитах , что обусловливает вариабельность картины заболевания.
Мутация DMPK приводит к нарушению гомеостаза ионов Ca++, а также процессов возбуждения и сокращения мышц, сердечной проводимости.
Рождение ребенка с конгенитальной формой DM1 часто является большой неожиданностью для семьи: во-первых, неожиданное сообщение врача (невролога, неонатолога) о рождении больного ребенка с наследственной патологией в семье, где родители считали себя здоровыми людьми, и разъяснение родственникам сути заболевания; во-вторых, неожиданность понимания того, что чаще всего больна и мать ребенка. Иногда диагноз устанавливается после смерти младенца или рождения мертвого ребенка. Эта реальная ситуация объясняется митохондриальным типом наследования DM1, не всегда подчиняющимся законам Менделя. Индивидуумы с умеренным фенотипом DM1 часто не осознают наличия у себя нервно-мышечного заболевания, которое, как правило, активно диагностируется только при выявлении в семье пациента с более тяжелым течением болезни. Это часто происходит, когда мать с асимптомным течением DM1, имеющая размер тринуклеотидного повтора CTG < 100, рождает младенца с врожденной (конгенитальной) DM1 с длиной CTG-повтора в тысячи.
КЛИНИЧЕСКАЯ ХАРАКТЕРИСТИКА И ДИАГНОСТИКА ДИСТРОФИЧЕСКОЙ МИОТОНИИ
Для миотонии Куршманна — Баттена — Штейнерта— Россолимо характерно сочетание миотонического синдрома, сердечно-сосудистых нарушений, эндокринной патологии.
1)Миотония — проявляется спазмами жевательных мышц и сгибателей кистей рук, характерны атрофические изменения в разных дистрофикгруппах мышц. Постепенно происходит угасание миотонических проявлений и прогрессирования мышечной дистрофии (атрофии), вследствие чего становится характерным вид пациента: голова опущена на грудь, лицо гипомимичное (Атрофии мимических мышц настолько стереотипны по виду, что все больные выглядят похожими: лицо удлиненное и утонченное вследствие слабости височных и жевательных мышц; шея тонкая (лебединая») из-зa атрофии грудиноключично-сосцевидных мышц; веки и углы рта опущены, нижняя половина лица провисает, что делает выражение лица печальным (полуптоз), дистальные амиотрофии конечностей. Типичны «выеденные» стопы, «обезьяньи» кисти. Походка степпажная, иногда при атрофиях проксимальных групп мышц напоминает утиную. Мышечный тонус снижен, глубокие рефлексы рано угасают. Опасным является парез мышц гортани с нарушением глотания, а также слабость дыхательной мускулатуры, в результате возможны нарушения сна (диссомнию): ночное апноэ («проклятие Ундины») и/или гиперсомнию, развитие пневмонии.
2)Сердечнососудистые нарушения — нарушения ритма сердца, гипертрофические изменения левого желудочка, выявляемые на ЭКГ, застойная сердечная недостаточность.
3)Эндокринные расстройства (в основном затрагиваются половые функции) — уменьшение размеров половых органов, снижение сексуального влечения, у женщин — расстройства менструального цикла, ожирение.
4)Патологии ЖКТ: поражение гладкой мускулатуры желудочно-кишечного тракта (орофарингеальная дисфункция, эзофагеальная дисфагия, нарушение моторики желудочно-кишечного тракта, вплоть до кишечной непроходимости — псевдообструкции кишечной проходимости, нарушение функции сфинктера прямой кишки: мегаколон; желчнокаменная болезнь) При ДM может поражаться гладкая мускулатура всего пищеварительного тракта, включая пищевод , желудок, тонкий и толстый кишечник, анус. Пациентов могут беспокоить боли в животе, запоры или диарея. Кроме того, в патологический процесс часто вовлекаются гладкие мышцы желчного пузыря и желчевыводящих путей. У больных ДM вероятность дискинезии желчевыводящих путей и желчнокаменной болезни намного выше, чем в среднем в популяции. Пациенты могут предъявлять жалобы на дискомфорт после приема жирной и острой пищи, боли в правом подреберье.

